FDA 21 CFR Part 11 Explained
13 June, 2022What is FDA 21 CFR Part 11 Compliance and Why Does it Matter?
The question above is one that numerous medical device developers who want to access the US market must have asked themselves at one point. It’s very easy to be intimidated by the myriad of initials and schedules you’ll be confronted with as you try to break into this huge marketplace. We’ve compiled a guide to help everyone understand what FDA 21 CFR Part 11 is and what it means to your company.
What is CFR 21 Part 11?

As noted in The Code of Federal Regulations, Part 11 of Title 21 clearly sets out what an organization in the US must do to successfully implement an FDA compliant, digital Quality Management System covering e-signatures and electronic records as opposed to ‘wet signatures’ and paper-based documentation.
Any Life Sciences company that wants to sell its services or products within the US must be in compliance with the 21 CFR Part 11 Electronic Signatures and Electronic Records regulation. This includes systems used in the research, manufacture and distribution of products such as medical devices, pharmaceuticals, blood, tissue, vaccines, and other biological products.
As such, it’s critical that the professionals in charge of building these support systems used by numerous organizations have a clear understanding of 21 CFR 11 to help life science companies remain compliant. But before we look at why CFR 11 compliance matters and what is required, here’s a bit of history.
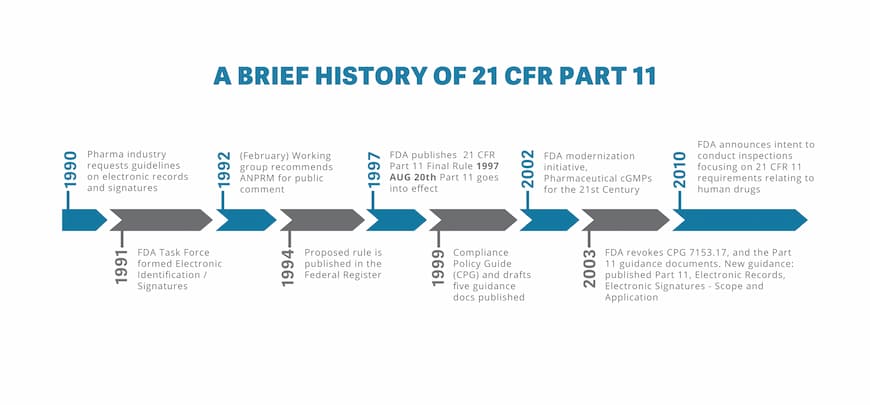
History of 21 CFR Part 11
At the request of the Pharma industry, the FDA introduced the 21 CFR Part 11 regulations in the early 1990s. Once Part 11 went into effect, there was a lot of variation in the approaches used for implementation and interpretation.
Not only were companies unclear on the specifics of implementing the requirements, they were also unsure about which records and systems were within the scope of Part 11. This led to the publication of the Compliance Policy Guide and other drafted Guidance Documents regarding Validation, Time Stamps, Glossary of Terms, Electronic Copies of Electronic Records & Maintenance of Electronic Records.
But in spite of the FDA’s efforts to give clear and consistent approaches, industries regulated by the FDA continue to express significant regulation-related concerns. The pharma industry, in particular, felt that compliance to Part 11 of the CFR:
- Failed to provide significant benefits to public health
- Discouraged technological advances and innovation due to fear of non-compliance
- Restricted use of technology
- Substantially increased the costs of using technology since systems required customization.
Modernizing Part 11
In 2002, a publication on Pharmaceutical cGMP was created to implement risk-based approaches and encourage adoption of new technological advances. The pharma industry was shocked to learn that the FDA revoked Enforcement policy CPG 7153.17 the following year, as well as five new guidance documents.
Fortunately, regulation 21 CFR Part 11 was still in effect with a new guidance titled Electronic Records; Electronic Signatures – Scope and Application being published in August 2003.
As the FDA’s standard for considering e-signatures and electronic records to be reliable and trustworthy, all regulated organizations need to care about Part 11. The regulation establishes minimums for the electronic equivalence to handwritten signatures and paper records. Bottom line is, you have to comply with Part 11 for FDA regulated activities if you want to get away from pen and paper.
Why Does 21 CFR 11 Compliance Matter?
As part of their response to the challenges and opportunities of the information age, the FDA released Part 11. Although lots of other industries were reaping the benefits of increased digitization, medical product developers were still wasting a lot of time collating paper documents to pass FDA audits and chasing real-world signatures.
21 CFR Part 11 not only addressed the need for heightened innovation in Pharma’s working methods, it also allowed new products to go to market faster with the aid of digital assistance. It also balanced this with the need to retain the highest level of control and authentication around approval processes for what are by all purposes and intent, potentially lethal products.
There are many reasons to pursue Part 11, not just because it’s a regulation. Below are some of the benefits of being FDA 21 CFR 11 compliant;
- Patient safety
- Product quality
- Operational consistency
- Protection and retrieval of electronic records
- Enable trending
- Minimize or eliminate management of paper documentation
- Enable faster data-related searches
- Electronic submission to the FDA
with the FDA, everyone benefits from CFR Part 11 which ensures the inspectability of e-records. The regulation ensures that just like paper records, the electronic records are always available for the retention period defined. What’s more, companies that move to electronic systems benefit from productivity, standardization, and efficiencies.
Similarly companies with e-records can provide better analysis of quality data trends making Part 11 also beneficial to the public. Products also appear on the market sooner since electronic submission for FDA approval of new products is faster than using paper or wet signatures.
Critical Requirements for 21 CFR Part 11
1: Validation
FDA’s 21 CFR Part 11 requires validation of systems to guarantee reliability, accuracy, and consistent intended performance. This means organizations should formally define how elements of their system are supposed to work, then create scripts and tests to validate the proper functionality. The process of validating your QMS might feel burdensome, but it demonstrates fitness for purpose and gives the regulator confidence that you can deliver to the required standards.
2: Data Integrity
According to Part 11, companies are required to have the digital controls and processes in place to ensure the integrity, authenticity, and confidentiality of electronic records.
The aim here is to make sure the information and data collated and shared is traceable, accurate, protected from loss or misuse, and fit for purpose. Imposing all the controls required by part 11 not only minimizes the risk of product failure, but also prevents harm to end users and costs of paying fines for compliance breaches and correcting mistakes.
3: Audit Trails
Organizations are required by Part 11 to have a complete version history available for every quality document in your system. To get complete accountability, and traceability over the decision making in the development process, you need to record every detail of every change and sign off event by date, time, and author.
4: Data Retrieval
Part 11 of FDA 21 CFR requires you to have the tools necessary to protect your document for accurate and ready retrieval throughout the retention process. Controlling these records means they are automatically indeed, archived, and available on demand. This will help support external audits by tracking all root causes of identified non-conformities in your system.
5: Security Controls
As specified under Part 11, companies can find all the controls they need over access and editing rights within their system. Included in the regulation are numerous exacting requirements to prevent accidental deletion or loss of data. This includes security breaches that can result in commercial failure, customer harm, and hefty regulator fines.
6: Electronic Signatures
Famously mapped out in Part 11 are all the requirements for the use of electronic signatures. 21 CFR 11 specifies that electronic signatures on documents must include date/time signature as applied, printed name of the signer, and intention of the e-signature as part of an evolving audit trail. But that’s not all; Part 11 made the authentication requirements for approval a lot more stringent in trying to match the legal confidence offered by a wet-signature. At the moment, it would be easier to falsify a pen and ink signature than an electronic one under the FDA rules.
7: Operational Controls
The use of operational systems check to enforce permitted sequencing of steps and events as appropriate is specified in Part 11. If you’re looking for more control over people and processes in the management of the development cycle, then setting up automated workflows for collection and approval of signatures will help. They ensure key documents are grouped together before review by specific individuals at specific moments. Part 11 brings clarity and order to complex processes and minimizes risks that could cause costly mistakes.
Final Word
FDA Regulated companies, need to meet very strict security and data integrity compliance requirements in the way they handle any kind of electronic records involving clinical data. That’s what 21 CFR 11 is about, and its ultimate objective is to protect the public.
So, in relation to the software that they use, the FDA needs convincing that the software is a secure closed system, and that it doesn’t mangle the data and doesn’t allow any tampering of the data either. Obviously since software is constantly evolving, one can never assure continued compliance from one release to the next, and anyone claiming they are compliant without a formal 21 CFR 11 validation process baked into the SQA release test procedure – like we do – is pulling a fast one.
To this end, to help our customers prepare for, and anticipate, an audit, ShareVault provides a 21 CFR 11 Compliance Validation Package which contains a risk assessment and all the necessary information about the new features in a release and their expected behavior, bugs fixed, test plans even a staging area when they can examine the operation of each new feature before it goes live.
Here is more information on ShareVault’s 21 CFR 11 Compliance